
Tüm iLive içeriği tıbbi olarak incelenir veya mümkün olduğu kadar gerçek doğruluğu sağlamak için kontrol edilir.
Sıkı kaynak bulma kurallarımız var ve yalnızca saygın medya sitelerine, akademik araştırma kurumlarına ve mümkün olduğunda tıbbi olarak meslektaş gözden geçirme çalışmalarına bağlanıyoruz. Parantez içindeki sayıların ([1], [2], vb.) Bu çalışmalara tıklanabilir bağlantılar olduğunu unutmayın.
İçeriğimizin herhangi birinin yanlış, güncel değil veya başka türlü sorgulanabilir olduğunu düşünüyorsanız, lütfen onu seçin ve Ctrl + Enter tuşlarına basın.
Pierre Robin sendromu
Makalenin tıp uzmanı

Son inceleme: 04.07.2025

Pierre Robin sendromu, tıpta Robin anomalisi olarak da bilinir, yüzün çene kısmının gelişiminde doğuştan gelen bir patolojidir. Hastalık adını, tüm belirtilerini ilk tanımlayan Fransız diş hekimi P. Robin'in onuruna almıştır. Lannelongue ve Menard, Pierre Robin sendromunu ilk olarak 1891'de mikrognati, yarık damak ve retroglossoptozisli iki hasta hakkındaki raporlarında tanımlamışlardır. Pierre-Robin, 1926'da klasik sendromun belirtilerini gösteren bir bebekte hastalığın bir vakasını yayınladı. 1974'e kadar, belirti üçlüsü Robin-Pierre sendromu olarak biliniyordu. Ancak, bu sendrom artık birden fazla anomalinin aynı anda bulunduğu malformasyonları tanımlamak için kullanılmaktadır.
Epidemioloji
8.500 canlı doğumda 1 prevalansı olan heterojen bir konjenital defekttir. X'e bağlı form hariç erkek-dişi oranı 1:1'dir.
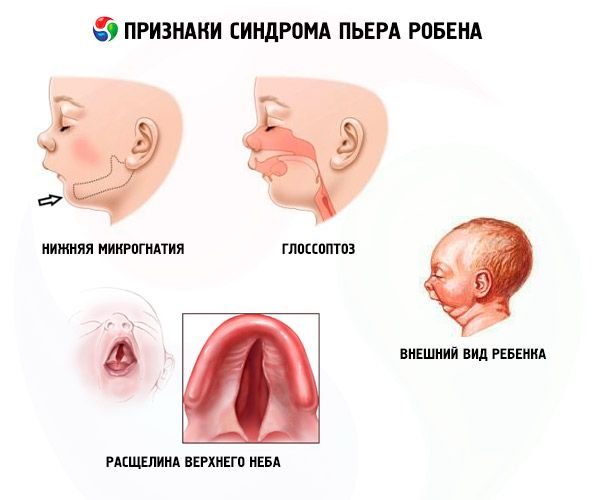
Bu hastaların %50'sinde tam olmayan yumuşak damak yarığı bulunurken, geri kalan kısmı kemerli ve alışılmadık derecede yüksek bir damakla, ancak yarık olmaksızın doğarlar.
Nedenler Pierre Robin sendromu
Hastalığın otozomal resesif kalıtım olasılığı düşünülüyor. Etiyolojiye bağlı olarak iki tip sendrom vardır: izole ve genetik olarak belirlenmiş. İzole tip, embriyonik gelişim sırasında çenenin alt kısmının sıkışması nedeniyle gelişir. Sıkıştırma şunlardan dolayı gelişebilir:
- Rahim içerisinde lokalize mühürlerin (kist, skar, tümör) varlığı.
- Çoğul gebelik.
Ayrıca fetüste çene gelişimi şu nedenlerle bozulabilir:
- Anne adayının hamilelik döneminde geçirdiği viral enfeksiyonlar.
- Nörotrofik bozukluklar.
- Gebe kadının vücudunda folik asit miktarının yetersiz olması.
Patogenez
Pierre Robin sendromu, doğum öncesi dönemde çok çeşitli patolojilerin neden olduğu embriyonik bozukluklar sonucu ortaya çıkar.
Pierre Robin sendromunun oluşumunu açıklayabilecek üç patofizyolojik teori vardır.
Mekanik Teori: Bu teori en olası olanıdır. Mandibular aparatın az gelişmesi gebeliğin 7. ve 11. haftaları arasında gerçekleşir. Dilin ağız boşluğunda yüksekte olması damakta yarık oluşumuna yol açar ve bu nedenle vena kava kapanmaz. Bu teori klasik ters U şeklinde yarık ve ilişkili dudak yarığının yokluğunu açıklar. Oligohidramnios etiyolojide rol oynayabilir çünkü amniyotik sıvının yokluğu çenenin deformasyonuna ve ardından dilin vena kava arasında sıkışmasına yol açabilir.
Nörolojik teori: Uvula ve faringeal kolon kaslarının elektromiyografisinde nörolojik gelişimde gecikme, hipoglossal sinirdeki iletim gecikmesine bağlı tat duyusunda gecikme olduğu görülmüştür.
Rombensefalonun Disnöroregülasyon Teorisi: Bu teori, ontogenez sırasında rombensefalonun gelişiminin bozulmasına dayanmaktadır.
Çocuğun alt çenesinin yetersiz gelişimi, ağız boşluğunun önemli ölçüde küçülmesine yol açar. Bu da, sözde psödomakroglossiye neden olur, yani dil, faringeal duvarın arkasına doğru yer değiştirir. Bu patoloji, hava yolu tıkanıklığının gelişmesine yol açar.
Bebek ağladığı veya hareket ettiği sürece hava yolu açık kalır, ancak bebek uykuya daldığında tekrar tıkanıklık oluşur.
Solunum bozuklukları nedeniyle bebeği besleme süreci çok zordur. Bu sırada neredeyse her zaman hava yolu tıkanıklığı meydana gelir. Tıbbi bir düzeltme uygulanmazsa, böyle bir patoloji tüm vücudun ciddi şekilde tükenmesine ve hatta ölüme yol açabilir.
Belirtiler Pierre Robin sendromu
Hastalığın üç ana belirtisi vardır:
- Alt mikrognati (alt çenenin az gelişmişliği, hastalığın %91.7'sinde görülür). Alt diş kemerinin üst kemerin 10-12 mm gerisine çekilmesiyle karakterizedir. Alt çene küçük bir gövdeye, geniş bir açıya sahiptir. Çocuk yaklaşık 5-6 yaşında normal gelişime ulaşır.
- Glossoptozis (Dilin yetersiz gelişmesi nedeniyle geriye çekilmesi; vakaların %70-85’inde görülür).
- Makroglossi ve ankiloglossi ise nispeten nadir görülen semptomlar olup vakaların %10-15’inde görülür.
- Gökyüzünde bir çatlak beliriyor.
- Bradipne ve dispne.
- Hafif siyanoz.
- Bebeği beslemeye çalışırken sıklıkla görülen asfiksi.
- Yutma imkânsızdır veya çok zordur.
- Kusma hissi.
- Vakaların %75’inde kulak anomalileri görülür.
- Hastaların %60’ında iletim tipi işitme kaybı görülürken, sadece %5’inde dış kulak yolu atrezisi, temporal kemiğin mastoid boşluğunun yetersiz pnömatizasyonu sonucu ortaya çıkar.
- İç kulak anomalileri (lateral semisirküler kanalların aplazisi, büyük vestibüler su kemeri, koklear tüy hücrelerinin kaybı).
- Burun malformasyonları nadir görülür ve çoğunlukla burun kökü anomalilerinden oluşur.
- Diş malformasyonları vakaların %30'unda görülür. Laringomalazi ve velofaringeal yetmezlik Pierre Robin sendromlu hastaların yaklaşık %10-15'inde görülür.
Pierre Robin sendromunun sistemik özellikleri
Kayıtlı vakaların %10-85’inde sistemik gelişimsel anomaliler tanımlanmaktadır.
Hastaların %10-30'unda göz anormallikleri görülür. Bunlara şunlar dahil olabilir: hipermetropi, miyopluk, astigmatizma, kornea sklerozu ve nazolakrimal kanal stenozu.
Kardiyovasküler patolojiler: iyi huylu kalp üfürümleri, pulmoner arter stenozu, patent duktus arteriosus, oval pencere, atriyal septal defekt ve pulmoner hipertansiyon. Prevalansları %5-58 arasında değişmektedir.
Kas-iskelet sistemiyle ilgili anomaliler (%70-80 vaka): sindaktili, displastik falankslar, polidaktili, klinodaktili, eklemlerin hipermobilitesi ve üst ekstremitelerin oligodaktili. Alt ekstremite anomalileri: ayak anomalileri (kulüp ayak, metatarsal adduksiyon), femoral malformasyonlar (valgus veya varus pelvis, kısa femurlar), kalça anomalileri (doğuştan çıkık, kontraktürler), diz eklemi anomalileri (GENU VALGUS, senkondroz). Omurga malformasyonları: skolyoz, kifoz, lordoz, vertebral displazi, sakrum ve koksigeal sinüs agenezisi.
Merkezi sinir sistemi patolojisi: epilepsi, sinir sisteminin gelişiminde gecikmeler, hidrosefali. Merkezi sinir sistemi defektlerinin sıklığı yaklaşık %50'dir.
Genitoüriner anomaliler: İnmemiş testis (%25), hidronefroz (%15) ve hidrosel (%10).
İlişkili sendromlar ve durumlar: Stickler sendromu, trizomi 11q sendromu, trizomi 18, 4q delesyon sendromu, romatoid artropati, hipokondroplazi, Moebius sendromu.
Aşamaları
Hastalığın şiddeti, çocuğun solunum yollarının durumuna bağlı olarak üç aşamadan oluşur:
- Hafif - beslenmede ufak sorunlar var, ancak nefes alma neredeyse hiç zor değil. Tedavi ayakta tedavi bazında gerçekleştirilir.
- Orta – nefes alma orta derecede zordur, çocuğu besleme orta derecede zordur. Tedavi hastanede yapılır.
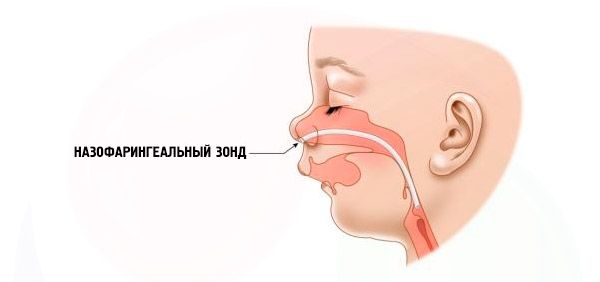
- Şiddetli – nefes almak çok zordur, çocuk normal şekilde beslenemez. Özel cihazlar (intranazal prob) kullanmak gerekir.
Komplikasyonlar ve sonuçları
Mikrognati ve glossoptozisin bir arada bulunması ciddi solunum komplikasyonlarına ve çocuğun beslenmesi sırasında sorunlara yol açabilir.
Pierre Robin sendromu aşağıdaki komplikasyonlara neden olur:
- Hava yolu tıkanıklığına bağlı hırıltılı solunum. Laringomalazi veya hatta uyku asfiksi.
- Çocuğun psikomotor gelişimi yaşıtlarından çok geridedir.
- Fiziksel gelişim de geri kalıyor.
- Hastaların konuşması bozulmuştur.
- Kronikleşip işitme kaybına yol açan sık kulak enfeksiyonları.
- Obstrüktif uyku apne sendromunda uykuda ölüm görülme sıklığı %14-91 arasında değişmektedir.
- Diş sorunları.
Teşhis Pierre Robin sendromu
Pierre Robin sendromunun teşhisi zor değildir. Klinik bulgulara dayanır. Diğer patolojileri dışlamak için bir genetikçiye danışmak çok önemlidir.
Robin'in doğuştan anomalisi olan çocuklarda dilin sürekli geriye doğru batması nedeniyle doğumdan itibaren solunum sorunları görülür. Bebek huzursuzdur, cildi mavimsidir, nefes alırken göğüsten hırıltı gelir. Beslenme sırasında boğulma meydana gelebilir. Tanı ayrıca çocuğun alışılmadık görünümüyle de konulabilir - "kuş yüzü". Hastalarda sıklıkla başka kusurlar da gelişir: miyopi, katarakt, genitoüriner sistem patolojisi, kalp patolojisi, omurganın gelişiminde anomaliler.
Bu klinik bulgulara dayanarak uzmanın doğru tanıyı koyması zor olmayacaktır.
Kim iletişim kuracak?
Tedavi Pierre Robin sendromu
Pierre Robin sendromlu bir çocuğun doğumundan hemen sonra tedavi yapılır. Hastalık hafifse, hastanın durumunu iyileştirmek için çocuğu sürekli olarak dikey veya karın üstü yatırmak gerekir. Bebeğin başı göğse doğru eğik olmalıdır. Beslenme sırasında, yemeğin solunum yoluna girmemesi için çocuğu yatay pozisyonda tutmanız önerilmez.
Alt çenenin az gelişmişliği oldukça belirginse, geri çekilen dili normal fizyolojik pozisyonuna getirmek için cerrahi müdahale kullanılır. Şiddetli vakalarda dil yukarı çekilir ve alt dudağa sabitlenir. Çok şiddetli vakalarda, alt çenenin trakeostomisi, glossopeksi ve distraksiyon osteogenezisi yapılmalıdır.
Konservatif tedavi de uygulanmaktadır.
İlaçlar
Fenobarbital. Uyku hapı ve sakinleştirici bir ilaçtır, antikonvülsan etkiye sahiptir. Her tablet 100 ml fenobarbital içerir. Dozaj, hastalığın ciddiyetine ve çocuğun durumuna bağlı olduğundan bireyseldir. İlaç, karaciğer yetmezliği, hiperkinezi, anemi, miyasteni, porfiri, diabetes mellitus, depresyon ve bileşenlere karşı intoleransı olan hastalar için yasaktır. Alırken aşağıdaki semptomlar mümkündür: baş dönmesi, asteni, halüsinasyonlar, agranülositoz, mide bulantısı, düşük tansiyon ve alerjiler.
Klonazepam. Epilepsi tedavisi için reçete edilen bir ilaç. İlaç, benzodiazepin türevi olan klonazepam aktif maddesini içerir. Antikonvülsan, anksiyolitik ve kas gevşetici etkileri vardır. Doz, ilgili hekim tarafından belirlenir, ancak maksimum - günde 250 mcg'yi geçmemelidir. Uykusuzluk, kas hipertonisi, psikomotor ajitasyon, panik bozuklukları durumunda almayın. Alırken aşağıdaki belirtiler mümkündür: uyuşukluk, mide bulantısı, dismenore, baş ağrısı, lökopeni, idrar retansiyonu veya inkontinans, alopesi, alerji.
Sibazon. Çözelti ve rektal tablet formunda mevcuttur. Etkin madde bir benzodiazepin türevidir (sibazon). Sedatif, anksiyolitik, antikonvülsan etkiye sahiptir. Dozaj bireyseldir. Kronik hiperkapni, miyasteni, benzodiazepin intoleransı olan hastaların ilacı almaları yasaktır. İlacı kullanırken aşağıdaki semptomlar gelişebilir: mide bulantısı, kabızlık, baş ağrısı, baş dönmesi, hıçkırık, idrar tutamama, alerjiler.
Cortexin liyofilizat. Nootropik etkiye sahip bir ilaç. İlaç suda çözünen polipeptit fraksiyonları ve glisin kompleksi içerir. Dozaj bireyseldir ve hastanın durumuna göre ilgili hekim tarafından reçete edilir. Cortexin'e karşı intoleransı olan hastaların ilacı almaları yasaktır. İlaç alerjik reaksiyonlara neden olabilir.
Fizyoterapi tedavisi
Sendromun hafif evrelerinde genellikle pozisyonel terapi uygulanır; çocuk, yer çekimi alt çenenin doğru şekilde büyümesini zorlayana kadar dik pozisyonda karnı üzerine yerleştirilir.
Cerrahi tedavi
Cerrahi tedavi öncelikle glossoptozisi düzeltmek için kullanılır. Birkaç yöntem vardır:
- Dilin gümüş bir iplikle desteklenmesi. İplik diş etinin alt kısmından ve alt dudaktan geçirilir. Yönteme Douglas denir.
- Duhamel yöntemi - hastanın dilinin tabanından ve her iki yanağından kalın bir gümüş iplik geçirilir. Otuz günden uzun süre kullanmayın.
- Dil uzatma ve fiksasyonu için ortopedik cihazlar.
- Bir yaşına gelindiğinde damak yarığını düzeltmek için ameliyat yapılabilir.
Tahmin
Hastalığın prognozu ve seyri şiddetlidir. Ölüm çoğunlukla yaşamın ilk günlerinde, hastalığın orta ve şiddetli evresinde gerçekleşir (nedeni asfiksidir). Ayrıca çok sayıda enfeksiyon nedeniyle ilk yılda ölüm riski oldukça yüksektir.
İki yaşın üzerindeki hastalarda prognoz iyidir.
 [ 36 ]
[ 36 ]