
Tüm iLive içeriği tıbbi olarak incelenir veya mümkün olduğu kadar gerçek doğruluğu sağlamak için kontrol edilir.
Sıkı kaynak bulma kurallarımız var ve yalnızca saygın medya sitelerine, akademik araştırma kurumlarına ve mümkün olduğunda tıbbi olarak meslektaş gözden geçirme çalışmalarına bağlanıyoruz. Parantez içindeki sayıların ([1], [2], vb.) Bu çalışmalara tıklanabilir bağlantılar olduğunu unutmayın.
İçeriğimizin herhangi birinin yanlış, güncel değil veya başka türlü sorgulanabilir olduğunu düşünüyorsanız, lütfen onu seçin ve Ctrl + Enter tuşlarına basın.
Huntington hastalığı
Makalenin tıp uzmanı

Son inceleme: 05.07.2025
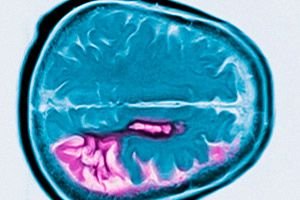
Huntington hastalığı, orta yaşta başlayan ilerleyici bilişsel gerileme, istemsiz hareketler ve bozulmuş motor koordinasyonuyla karakterize otozomal dominant nörodejeneratif bir hastalıktır. Tanı genetik testlerle doğrulanır. Tedavi öncelikle semptomatiktir. Kan bağı olan akrabalar için genetik test önerilebilir. George Huntington, Long Island sakinlerinde bir aile vakasını inceledikten sonra durumu ilk olarak 1872'de tanımladı.
Huntington hastalığının görülme sıklığı 100.000 kişide yaklaşık 10 vakadır ve geç başlangıçlı olması nedeniyle 100.000 kişiden yaklaşık 30'unun yaşamları boyunca bu hastalığa yakalanma riski %50'dir. Hastalık çoğunlukla 35 ila 40 yaşları arasında ortaya çıksa da başlangıç yaş aralığı oldukça geniştir; en erken başlangıç 3 yaşında, en geç başlangıç ise 90 yaşındadır. Başlangıçta hastalığın %100 penetransa sahip olduğu düşünülüyordu ancak artık bunun her zaman böyle olmadığına inanılıyor. Hastalığın genini babasından alan bireylerde hastalık, patolojik geni annesinden alan bireylere göre ortalama 3 yıl daha erken ortaya çıkar. Patolojik geni babasından alan hastaların yaklaşık %80'inde hastalık 20 yaşından önce ortaya çıkar. Yavrularda genetik bir kusurun daha erken ortaya çıkması olgusuna antisipasyon denir.
 [ 1 ]
[ 1 ]
Huntington hastalığına ne sebep olur?
Huntington hastalığının cinsiyet tercihi yoktur. Küçük nöronların dejenerasyona uğradığı ve nörotransmitterlerin - gama-aminobütirik asit (GABA) ve P maddesi - seviyesinin düştüğü kaudat çekirdeğinin atrofisi gösterilmiştir.
CAG (sistein-alanin-glisin) DNA dizilerinin aminoasit glutamin kodlayan sayısının ("genişleme") arttığı bir mutant gen, Huntington hastalığının gelişiminden sorumludur. Bu genin ürünü olan büyük protein huntingtin, bilinmeyen bir mekanizma ile hastalığa yol açan aşırı miktarda poliglutamin kalıntısı içerir. CAG tekrarları ne kadar fazlaysa, hastalık o kadar erken başlar ve seyri o kadar şiddetli olur. Nesilden nesile tekrar sayısı artabilir ve bu da zamanla aile fenotipinin kötüleşmesine yol açar.
Parkinson hastalığındaki genetik ve biyokimyasal değişikliklere yönelik önemli ilgiye rağmen, hastalık için bir gen arayışı 1970'lerin sonuna kadar başarısız oldu. O zamanlar Nancy Wexler ve Allan Tobin, Huntington hastalığı için bir gen bulma stratejisini tartışmak üzere Hereditary Disease Foundation tarafından desteklenen bir çalıştay düzenlediler. Toplantıya katılan David Housman, David Botstein ve Ray White, yakın zamanda geliştirilen rekombinant DNA tekniklerinin bu hedefe ulaşmaya yardımcı olabileceğini öne sürdüler. Projedeki temel görevlerden biri, DNA örnekleri elde etmek için Huntington hastalığı olan birçok nesile sahip büyük bir aile bulmaktı. 1979'da Venezuela ve Amerika Birleşik Devletleri'nden bilim insanlarının ortak projesi, Maracheibo Gölü (Venezuela) kıyılarında yaşayan Huntington hastalığı olan büyük bir aileyi incelemek için başlatıldı. 1983 yılında Huntington hastalığı geni 4. kromozomun kısa kolunun sonunda lokalize edildi (Gusella ve ark., 1983) ve on yıl sonra bu genin mutasyonunun sitozin-adenin-guanin (CAG) trinükleotidinin tekrar sayısındaki artıştan oluştuğu ortaya çıktı (Huntington Hastalığı İşbirliği Araştırma Grubu, 1993). Bu bilimsel grup tarafından geliştirilen metodoloji şu anda yeni genlerin pozisyonel klonlanması için standart olarak kabul edilmektedir.
Vahşi tip gen 10-28 CAG tekrarına sahipken, Huntington hastalığına neden olan genin mutant formu 39'dan 100'den fazla CAG tekrarına kadar artmış bir genişliğe sahiptir. Trinükleotid tekrarlarının genişlemesinin keşfi hastalığın birçok klinik özelliğini açıklamaya yardımcı olmuştur. Özellikle başlangıç yaşı ile tekrarlayan trinükleotidlerin bulunduğu bölgenin uzunluğu arasında ters bir korelasyon bulunmuştur. Paternal kalıtım beklentisi, tekrar sayısındaki artışın erkeklerde genellikle spermatogenez sırasında meydana gelmesi gerçeğiyle açıklanabilir. Yeni mutasyonların analizi, bunların genellikle ebeveynlerden birinin, genellikle babanın, CAG tekrar sayısının 28'den yüksek olması durumunda meydana geldiğini göstermiştir; bu durumda, bu tekrarların sayısı bir sonraki nesilde artmıştır. Artık tekrar sayısı 28'den fazla değilse, nesilden nesile stabil bir şekilde aktarıldığı belirlenmiştir. Tekrar sayısı 29 ila 35 arasındaysa, Huntington hastalığının belirtileri ortaya çıkmaz, ancak yavrulara aktarıldığında bu bölgenin uzunluğu artabilir. Tekrar sayısı 36 ila 39 arasındaysa, bazı durumlarda (ancak her zaman değil) hastalık klinik olarak kendini gösterebilir (eksik penetrans) ve yavrulara aktarıldığında trinükleotid tekrarlarının sayısında artış olabilir. Tekrar sayısı 40'ı aşarsa, hastalık hemen hemen tüm vakalarda görülür ve yavrulara aktarıldığında tekrarların daha da genişlemesi mümkündür. Tekrar sayısındaki artışın nedenleri bilinmemektedir.
Huntington hastalığının patomorfolojisi
Huntington hastalığı, esas olarak kaudat çekirdek ve putamende ve bir dereceye kadar da korteks ve diğer beyin yapılarında nöronal kayıpla karakterizedir. Huntington hastalığında toplam beyin ağırlığı sadece nöron sayısındaki azalmayla değil, aynı zamanda beyaz cevher kaybıyla da azalır. Serebral kortekste, V ve VI. katmanlardaki hücreler en çok etkilenir. Mikro ve makroskobik dejeneratif değişikliklerin şiddeti (ölüm yaşına göre ayarlanmış) CAG tekrarlarının sayısıyla ilişkilidir. Huntington hastalığının birkaç yüz vakasındaki değişikliklerin ayrıntılı patolojik analizi, striatum dejenerasyonunun kaudat çekirdeğin dorsomedial kısmında ve putamenin dorsolateral kısmında başladığını ve daha sonra ventrale doğru yayıldığını göstermiştir. Kaudat çekirdek ve putamendeki farklı nöron grupları farklı derecelerde etkilenir. Striatumdaki internöronlar nispeten sağlam kalır, ancak bazı projeksiyon nöronları seçici olarak etkilenir. Huntington hastalığının juvenil formunda striatumdaki patolojik değişiklikler daha belirgin ve daha yaygındır; serebral korteks, serebellum, talamus ve globus pallidusu içerir.
Huntington hastalığında nörokimyasal değişiklikler
GABA. Huntington hastalığı olan hastalarda beynin nörokimyasal çalışmaları, striatumdaki GABA konsantrasyonunda önemli bir azalma olduğunu ortaya koydu. Sonraki çalışmalar, Huntington hastalığının GABAerjik nöronların sayısındaki azalma ile ilişkili olduğunu doğruladı ve GABA konsantrasyonlarının sadece striatumda değil, aynı zamanda projeksiyon bölgelerinde de - globus pallidus'un dış ve iç segmentleri ve substantia nigra'da - azaldığını gösterdi. Huntington hastalığı olan beyinde, reseptör bağlanma çalışmaları ve mRNA'nın in situ hibridizasyonu kullanılarak GABA reseptörlerindeki değişiklikler de tespit edildi. GABA reseptörlerinin sayısı kaudat çekirdekte ve putamende orta derecede azaldı, ancak substantia nigra'nın retiküler kısmında ve globus pallidus'un dış segmentinde muhtemelen sinir uyarımına bağlı aşırı duyarlılıktan kaynaklanan bir artış görüldü.
Asetilkolin. Asetilkolin, striatumdaki büyük dikensiz internöronlar tarafından bir nörotransmitter olarak kullanılır. Huntington hastalığı olan hastalarda yapılan erken postmortem çalışmalar, striatumdaki kolin asetiltransferaz (ChAT) aktivitesinin azaldığını gösterdi ve bu da kolinerjik nöronların kaybını düşündürdü. Ancak, GABAerjik nöronlardaki önemli azalmayla karşılaştırıldığında, kolinerjik internöronlar nispeten korunmuştur. Bu nedenle, striatumdaki asetilkolinesteraz pozitif nöronların yoğunluğu ve ChAT aktivitesi, yaşa eşleştirilmiş kontrollerle karşılaştırıldığında aslında nispeten yüksektir.
Madde P. Madde P, striatumun birçok orta dikenli nöronunda bulunur ve bunlar baskın olarak globus pallidus'un iç segmentine ve substantia nigra'ya projekte olur ve genellikle dinorfin ve GABA da içerir. Huntington hastalığında striatum ve substantia nigra'nın pars retikülaris'indeki Madde P seviyeleri azalır. Hastalığın terminal evresinde, immünohistokimyasal çalışmalar, madde P içeren nöron sayısında önemli bir azalma olduğunu ortaya koymuştur. Daha erken evrelerde, madde P içeren ve globus pallidus'un iç segmentine projekte olan nöronlar, substantia nigra'nın pars retikülaris'ine projekte olan nöronlara kıyasla nispeten korunur.
Opioid peptitler. Enkefalin, dolaylı yolun orta dikenli projeksiyon GABAerjik nöronlarında bulunur ve bunlar globus pallidusun dış segmentine projeksiyon yapar ve D2 reseptörleri taşır. İmmünohistokimyasal çalışmalar, globus pallidusun dış segmentine projeksiyon yapan enkefalin içeren nöronların Huntington hastalığında erken dönemde kaybolduğunu göstermiştir. Bu hücreler, globus pallidusun iç segmentine projeksiyon yapan P maddesi içeren hücrelerden daha erken ölüyor gibi görünüyor.
Katekolaminler. Biyojenik aminler (dopamin, serotonin) içeren ve striatuma projeksiyon yapan nöronlar, substantia nigra'nın kompakt kısmında, ventral tegmentumda ve raphe çekirdeklerinde yer alır. İnsan striatumuna noradrenerjik projeksiyonlar minimal olsa da, striatumdaki serotonin ve dopamin seviyeleri (doku gramı başına) yüksektir ve bu, striatumun kendi nöronlarının belirgin kaybına rağmen bu afferent projeksiyonların korunduğunu gösterir. Substantia nigra'nın dopaminerjik nöronları, Huntington hastalığının hem klasik hem de juvenil formlarında sağlam kalır.
Somatostatin/nöropeptid Y ve nitrik oksit sentaz. Huntington hastalığında striatumdaki somatostatin ve nöropeptid Y seviyelerinin ölçümü normal dokulara kıyasla 4-5 kat artış olduğunu gösterdi. İmmünohistokimyasal çalışmalar nöropeptid Y, somatostatin ve nitrik oksit sentaz içeren striatal internöronların mutlak korunduğunu gösterdi. Bu nedenle, bu nöronlar patolojik sürece dirençlidir.
Uyarıcı amino asitler. Huntington hastalığında seçici hücre ölümünün glutamat kaynaklı nörotoksik etkiden kaynaklandığı öne sürülmüştür. Huntington hastalığının striatumunda glutamat ve kinolinik asit (serotonin metabolizmasının bir yan ürünü ve glutamat reseptörlerinin bir agonisti olan endojen bir nörotoksin) seviyeleri hafifçe değişmiştir, ancak MR spektroskopisi kullanılarak yapılan yakın tarihli bir çalışma, glutamat seviyelerinde canlıda bir artış olduğunu ortaya koymuştur. Huntington hastalığının striatumunda kinolinik asit sentezinden sorumlu glial enzimin seviyesi normale göre yaklaşık 5 kat artarken, kinolinik asidin parçalanmasını sağlayan enzimin aktivitesi Huntington hastalığında sadece %20-50 oranında artmıştır. Bu nedenle, Huntington hastalığında kinolinik asit sentezi artmış olabilir.
Huntington hastalığında uyarıcı amino asit (EAA) reseptörleri üzerine yapılan çalışmalar, striatumdaki NMDA, AMPA, kainat ve metabotropik glutamat reseptörlerinin sayısında ve serebral korteksteki AMPA ve kainat reseptörlerinde önemli bir azalma olduğunu ortaya koymuştur. Huntington hastalığının geç evresinde NMDA reseptörleri neredeyse yokken, klinik öncesi ve erken evrelerde bu reseptörlerin sayısında önemli bir azalma olduğu kaydedilmiştir.
Seçici duyarlılık. Huntington hastalığında, belirli tipteki striatal hücreler seçici olarak kaybolur. Globus pallidusun dış segmentine projeksiyon yapan ve GABA ve enkefalin içeren orta dikenli nöronlar, GABA ve substantia nigranın retiküler kısmına projeksiyon yapan nöronlar gibi hastalığın çok erken dönemlerinde ölür. GABA ve enkefalin içeren ve globus pallidusun dış segmentine projeksiyon yapan nöronların kaybı bu yapıyı ortadan kaldırır ve bu da subtalamik çekirdeğin aktif inhibisyonuna yol açar. Subtalamik çekirdeğin azalmış aktivitesi, Huntington hastalığında görülen koreiform hareketleri açıklayabilir. Subtalamik çekirdeğin fokal lezyonlarının koreye neden olabileceği uzun zamandır bilinmektedir. GABA ve substantia nigra pars retikülarise projeksiyon yapan P maddesi nöronlarının kaybı, Huntington hastalığında görülen okülomotor bozukluklardan sorumlu olabilir. Bu yol normalde, üst kollikulusa projeksiyon yapan substantia nigra pars retikülaris nöronlarını engeller ve bu da sakadları düzenler. Juvenil Huntington hastalığında, yukarıda belirtilen yollar daha ciddi şekilde etkilenir ve ayrıca, globus pallidus'un iç segmentine olan striatal projeksiyonlar erken kaybolur.
Huntington hastalığına neden olan mutasyona uğrayan gen tarafından kodlanan huntingtin proteini, beynin ve diğer dokuların çeşitli yapılarında bulunur. Huntingtin normalde nöronların sitoplazmasında baskın olarak bulunur. Protein beyindeki çoğu nöronda bulunur, ancak son veriler içeriğinin matris nöronlarında striozomal nöronlardan daha yüksek ve projeksiyon nöronlarında internöronlardan daha yüksek olduğunu göstermektedir. Bu nedenle nöronların seçici duyarlılığı, normalde belirli nöronal popülasyonlarda bulunan huntingtin içerikleriyle ilişkilidir.
Huntington hastalığı olan hastaların beyinlerinde olduğu gibi, Huntington hastalığı geninin N-terminal parçası için transgenik olan ve tekrar sayısı artmış farelerde, huntingtin nöronların çekirdeklerinde yoğun kümeler oluşturur. Bu intranükleer inklüzyonlar striatal projeksiyon nöronlarında oluşur (ancak internöronlarda değil). Transgenik farelerde, inklüzyonlar semptomların başlamasından birkaç hafta önce oluşur. Bu veriler, inklüzyonları trinükleotid tekrarlarını veya bir parçasını kodlayan artmış sayıda glutamin kalıntısı içeren huntingtin proteininin çekirdekte biriktiğini ve sonuç olarak hücresel işlevler üzerindeki kontrolünü bozabileceğini göstermektedir.
Huntington hastalığının belirtileri
Huntington hastalığı olan hastalarda ilk semptomların ortaya çıktığı yaşı kesin olarak belirlemek zordur, çünkü hastalık kendini kademeli olarak gösterir. Kişilik ve davranışta değişiklikler, hafif koordinasyon bozuklukları, daha belirgin semptomların ortaya çıkmasından yıllar önce ortaya çıkabilir. Tanı konulduğunda, çoğu hastada koreik hareketler, ince hareketlerin koordinasyonunda bozulma ve istemli göz hareketlerinin yavaş oluşumu görülür. Hastalık ilerledikçe aktivitelerini organize etme yeteneği bozulur, hafıza azalır, konuşma zorlaşır, okülomotor bozukluklar ve koordineli hareketlerin bozulmuş performansı artar. Hastalığın erken evresinde kas ve duruşta herhangi bir değişiklik olmasa da, ilerledikçe distonik duruşlar gelişebilir ve zamanla baskın bir semptoma dönüşebilir. Geç evrede konuşma peltekleşir, yutma önemli ölçüde zorlaşır, yürüme imkansız hale gelir. Huntington hastalığı genellikle 15-20 yıl içinde ilerler. Terminal evrede hasta çaresizdir ve sürekli bakıma ihtiyaç duyar. Ölümcül sonuç doğrudan birincil hastalıkla değil, zatürre gibi komplikasyonlarıyla ilgilidir.
Huntington hastalığında bunama
ICD-10 kodu
P02.2. Huntington hastalığında demans (G10).
Demans, beynin striatal sistemine ve diğer subkoekal çekirdeklere baskın hasar veren sistemik dejeneratif-atrofik bir sürecin tezahürlerinden biri olarak gelişir. Otozomal dominant bir şekilde kalıtılır.
Hastalık genellikle yaşamın üçüncü veya dördüncü on yılında koreoform hiperkinezi (özellikle yüz, kollar, omuzlar, yürüyüşte), kişilik değişiklikleri (heyecanlı, histerik ve şizoid tip kişilik anomalileri), psikotik bozukluklar (kasvetlilik, somurtkanlık, disfori ile birlikte görülen özel depresyon; paranoid ruh hali) ile kendini gösterir.
Tanı için özellikle önemli olan koreoform hiperkinezi, demans ve kalıtsal yükün birleşimidir. Aşağıdakiler bu demans için özeldir:
- yavaş ilerleme (ortalama 10-15 yıl): üretken zihinsel çalışma gerektiren durumlarda (kavramsal düşünme, yeni şeyler öğrenme) kendine bakma yeteneğinin geri kalanı ile belirgin entelektüel yetersizlik arasındaki ayrışma;
- dikkat dağınıklığı ve hastanın tutumlarındaki tutarsızlığa dayanan belirgin zihinsel performans dengesizliği (hiperkinezi benzeri "sarsıntılı" düşünme);
- yüksek kortikal fonksiyonların belirgin ihlallerinin atipikliği;
- Demans artışı ile psikotik bozuklukların şiddeti arasında ters ilişki vardır.
Hastalığın klinik tablosunda psikotik (paranoyak kıskançlık, zulüm sanrıları) ve disforik bozuklukların yüksek oranda bulunması dikkate alınarak tedavide, dopaminerjik reseptörleri bloke eden (fenotiyazin ve butirofenon türevleri) veya dokulardaki dopamin düzeyini düşüren (reserpin) çeşitli nöroleptikler kullanılır.
Haloperidol (2-20 mg/gün), tiaprid (100-600 mg/gün) en fazla üç ay süreyle, tiyoridazin (100 mg/güne kadar), reserpin (0,25-2 mg/gün) ve antikonvülsan klonazepam (1,5-6 mg/gün) kullanılır. Bu ilaçlar hiperkineziyi azaltmaya, duygusal gerginliği yumuşatmaya ve kişilik bozukluklarını telafi etmeye yardımcı olur.
Ruhsal bozuklukların yatarak tedavisi, hastanın önde gelen sendromu, yaşı ve genel durumu dikkate alınarak gerçekleştirilir. Ayakta tedavide, tedavi prensipleri aynıdır (hareket bozukluklarının sürekli idame tedavisi, ilacın periyodik olarak değiştirilmesi). Ayakta tedavide daha düşük dozlarda nöroleptikler kullanılır.
Hafif ve orta dereceli demans için rehabilitasyon önlemleri arasında mesleki terapi, psikoterapi ve bilişsel eğitim yer alır. Aile üyeleriyle çalışmak ve hastaya bakan kişilere psikolojik destek sağlamak gerekir. Hastalık önlemenin temel yöntemi, çocuk sahibi olmaya karar verirken DNA analizi için bir yönlendirme ile hastanın en yakın akrabalarına tıbbi ve genetik danışmanlık yapılmasıdır.
Prognoz genellikle elverişsizdir. Hastalığın seyri yavaş ilerler ve hastalık genellikle 10-15 yıl sonra ölüme yol açar.
[ 18 ]
Seni rahatsız eden nedir?
Huntington hastalığının tedavisi
Huntington hastalığının tedavisi semptomatiktir. Kore ve ajitasyon nöroleptiklerle (örneğin, günde 3 kez oral olarak 25-300 mg klorpromazin, günde 2 kez oral olarak 5-45 mg haloperidol) veya günde bir kez oral olarak 0,1 mg rezerpinle kısmen bastırılabilir. Dozlar tolere edilebilen maksimum seviyeye çıkarılır (uyuşukluk, parkinsonizm gibi yan etkiler ortaya çıkmadan önce; rezerpin için hipotansiyon). Ampirik tedavinin amacı, Nmetil-O-aspartat reseptörleri aracılığıyla glutamatergik iletimi azaltmak ve mitokondride enerji üretimini sürdürmektir. Beyindeki GABA'yı artırmayı amaçlayan tedavi etkisizdir.
Genetik test ve danışmanlık önemlidir çünkü hastalığın belirtileri doğurganlık yıllarından sonra ortaya çıkar. Pozitif aile geçmişi olan kişiler ve test yaptırmakla ilgilenenler, tüm etik ve psikolojik etkiler göz önünde bulundurularak uzmanlaşmış merkezlere yönlendirilir.
Huntington hastalığının semptomatik tedavisi
Huntington hastalığının ilerlemesini durdurabilecek etkili bir tedavi yoktur. Çeşitli ilaçların birkaç denemesi yürütülmüştür, ancak önemli bir etki elde edilememiştir. Nöroleptikler ve diğer dopamin reseptör antagonistleri, Huntington hastalığı olan hastalarda zihinsel bozuklukları ve istemsiz hareketleri düzeltmek için yaygın olarak kullanılır. İstemsiz hareketler, dopaminerjik ve GABAerjik sistemler arasındaki dengesizliği yansıtır. Buna göre, nöroleptikler aşırı dopaminerjik aktiviteyi azaltmak için kullanılır. Ancak, bu ilaçların kendileri önemli bilişsel ve ekstrapiramidal yan etkilere neden olabilir. Ayrıca, hastanın psikoz veya ajitasyon geliştirdiği durumlar dışında, etkinlikleri kanıtlanmamıştır. Nöroleptikler genellikle disfajiye veya diğer hareket bozukluklarına neden olur veya bunları şiddetlendirir. Risperidon, klozapin ve olanzapin gibi yeni nesil nöroleptikler, daha az ekstrapiramidal yan etkiye neden olmalarına rağmen paranoid semptomları veya artan sinirliliği azaltabildikleri için Huntington hastalığının tedavisinde özellikle yararlı olabilir.
Tetrabenazin ve rezerpin de dopaminerjik sistemin aktivitesini azaltır ve hastalığın erken evrelerinde istemsiz hareketlerin şiddetini azaltabilir. Ancak bu ilaçlar depresyona neden olabilir. Hastalığın kendisi sıklıkla depresyona neden olduğundan, bu yan etki rezerpin ve tetrabenazin kullanımını önemli ölçüde sınırlar. Hastalığın geç evrelerinde, dopamin reseptörleri taşıyan hücreler ölür, bu nedenle dopamin reseptör antagonistlerinin etkinliği zayıflar veya kaybolur.
Nöroleptikler, antidepresanlar ve anksiyolitikler, Huntington hastalığı olan hastalarda psikoz, depresyon ve sinirlilik tedavisinde kullanılır, ancak bunlar yalnızca hasta gerçekten bu semptomları yaşadığı sürece reçete edilmelidir. Hastalığın bir aşamasında yardımcı olabilecek ilaçlar, hastalık ilerledikçe etkisiz hale gelebilir veya hatta zararlı olabilir.
Huntington hastalığı olan hastalarda GABA reseptör agonistleri test edilmiştir, çünkü Huntington hastalığının striatumdaki GABA seviyelerinde önemli bir azalma ve projeksiyon bölgelerindeki GABA reseptörlerinde aşırı duyarlılık olduğu gösterilmiştir. Benzodiazepinler, istemsiz hareketlerin ve bilişsel bozukluğun stres ve anksiyete ile ağırlaştığı durumlarda etkili olduğu kanıtlanmıştır. İstenmeyen sedatif etkilerden kaçınmak için bu ilaçların düşük dozları reçete edilmelidir. Huntington hastalığı olan hastaların çoğunda, ilaçların hiçbiri yaşam kalitesinde önemli bir iyileşmeye yol açmaz.
Parkinson semptomları olan erken başlangıçlı Huntington hastalığında dopaminerjik ajanlar denenebilir ancak bunların etkinliği sınırlıdır. Dahası, levodopa bu hastalarda miyoklonusa neden olabilir veya miyoklonusu artırabilir. Aynı zamanda, baklofen Huntington hastalığı olan bazı hastalarda rijiditeyi azaltabilir.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
Huntington hastalığının önleyici (nöroprotektif) tedavisi
Huntington hastalığındaki genetik kusur bilinmesine rağmen, bunun seçici nöronal dejenerasyona nasıl yol açtığı hala belirsizliğini korumaktadır. Oksidatif stresi ve eksitotoksisiteyi azaltmayı amaçlayan önleyici tedavilerin hastalığın ilerlemesini yavaşlatabileceği veya durdurabileceği varsayılmaktadır. Durum, genetik kusurun uzun yıllar bilinmediği ancak ikincil etki olan bakır birikimini hedefleyen önleyici tedavilerin "iyileştirildiği" hepatolentiküler dejenerasyona biraz benzeyebilir. Bu bağlamda, Huntington hastalığının enerji metabolizması bozukluğu ve eksitotoksisite nedeniyle hücre ölümüyle ilişkili olduğu hipotezi özellikle dikkat çekmiştir. Hastalığın kendisi, hücresel ve metabolik işlevleri bozan huntingtin'in N-terminal parçalarının intranükleer agregasyonu nedeniyle hücre ölümüne neden olabilir. Bu süreç, eksitotoksik hasara karşı daha yüksek duyarlılıkları nedeniyle bazı nöron gruplarını diğerlerinden daha fazla etkileyebilir. Bu durumda, uyarıcı amino asit reseptör antagonistleri veya serbest radikal hasarını önleyen ajanlarla önleyici tedavi, hastalığın başlangıcını ve ilerlemesini önleyebilir veya geciktirebilir. Amyotrofik lateral sklerozun laboratuvar modellerinde, antioksidan ajanların ve reseptör antagonistlerinin (RAA'lar) hastalığın ilerlemesini yavaşlatabildiği gösterilmiştir. Benzer yaklaşımlar Huntington hastalığında etkili olabilir. Glutamat reseptör antagonistleri ve mitokondriyal elektron taşıma zincirinin II. kompleksinin işlevini artıran ajanların klinik deneyleri şu anda devam etmektedir.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]