
Tüm iLive içeriği tıbbi olarak incelenir veya mümkün olduğu kadar gerçek doğruluğu sağlamak için kontrol edilir.
Sıkı kaynak bulma kurallarımız var ve yalnızca saygın medya sitelerine, akademik araştırma kurumlarına ve mümkün olduğunda tıbbi olarak meslektaş gözden geçirme çalışmalarına bağlanıyoruz. Parantez içindeki sayıların ([1], [2], vb.) Bu çalışmalara tıklanabilir bağlantılar olduğunu unutmayın.
İçeriğimizin herhangi birinin yanlış, güncel değil veya başka türlü sorgulanabilir olduğunu düşünüyorsanız, lütfen onu seçin ve Ctrl + Enter tuşlarına basın.
Yeni bulgular Rett sendromunun nedenlerinin daha iyi anlaşılmasına katkıda bulunuyor
Son inceleme: 02.07.2025

Rett sendromu, şu anda bir tedavisi veya iyi bir tedavisi olmayan nadir bir nörogelişimsel bozukluktur. Birçoğu otizm spektrum bozukluklarıyla örtüşen şiddetli fiziksel ve bilişsel semptomlara neden olur.
Rett sendromu, beyinde yüksek oranda ifade edilen ve nöronların sağlığını korumada önemli bir rol oynadığı görülen MECP2 genindeki mutasyonlardan kaynaklanır. Gen, X kromozomunda bulunur ve sendrom öncelikle kızları etkiler. Rett sendromu için tedaviler geliştirmek amacıyla araştırmacılar, MECP2'yi ve beyindeki işlevlerini daha iyi anlamak istiyor.
Whitehead Enstitüsü kurucu ortağı Rudolf Jaenisch de dahil olmak üzere araştırmacılar, MECP2'yi onlarca yıldır inceliyorlar, ancak gen hakkında birçok temel gerçek hala bilinmiyor. Gen tarafından kodlanan protein, MECP2, gen düzenlemesinde rol oynar; DNA'ya bağlanır ve çeşitli diğer genlerin ifade seviyelerini veya ürettikleri protein miktarını etkiler.
Ancak araştırmacıların elinde MECP2'den etkilenen genlerin tam bir listesi yoktu ve MECP2'nin bu genleri nasıl etkilediği konusunda bir fikir birliği yoktu.
MECP2'nin erken dönem çalışmaları, bunun bir baskılayıcı olduğunu ve hedef genlerinin ifadesini azalttığını ileri sürmüştü ancak Jaenisch ve diğerlerinin araştırmaları daha önce MECP2'nin aynı zamanda bir aktivatör olarak hareket ettiğini ve hedeflerinin ifadesini artırdığını göstermişti - ve ilk etapta bir aktivatör olabilirdi. Ayrıca MECP2'nin etki mekanizması veya proteinin gen ifadesinde değişikliklere neden olmak için tam olarak ne yaptığı bilinmiyordu.
Teknolojideki sınırlamalar araştırmacıların bu sorulara açıklık getirmesini engelledi. Ancak Yanish, laboratuvarının doktora sonrası araştırmacısı Yi Liu ve Yanish'in eski laboratuvar üyesi, şu anda Université de Montréal'deki CHU Sainte-Justine araştırma merkezinde yardımcı doçent olan Anthony Flamier, MECP2 hakkında kalan bu soruları yanıtlamak ve beyin sağlığı ve hastalıklarındaki rolüne dair yeni içgörüler elde etmek için son teknoloji teknikleri kullandılar.
Bulguları Neuron dergisinde yayımlandı ve araştırmacılar ayrıca diğer araştırmacılar için bir kaynak olması amacıyla MECP2 verilerinin çevrimiçi deposu olan MECP2-NeuroAtlas portalını oluşturdular.
"Bu makalenin, insanların MECP2'nin Rett sendromuna nasıl sebep olduğuna dair anlayışını kökten değiştireceğini düşünüyorum. Mekanizma hakkında tamamen yeni bir anlayışa sahibiz ve bu, hastalık için tedaviler geliştirmek için yeni yollar sağlayabilir," diyor MIT'de biyoloji profesörü olan Janisch.
Beyindeki MECP2'nin Daha Derin Anlaşılması
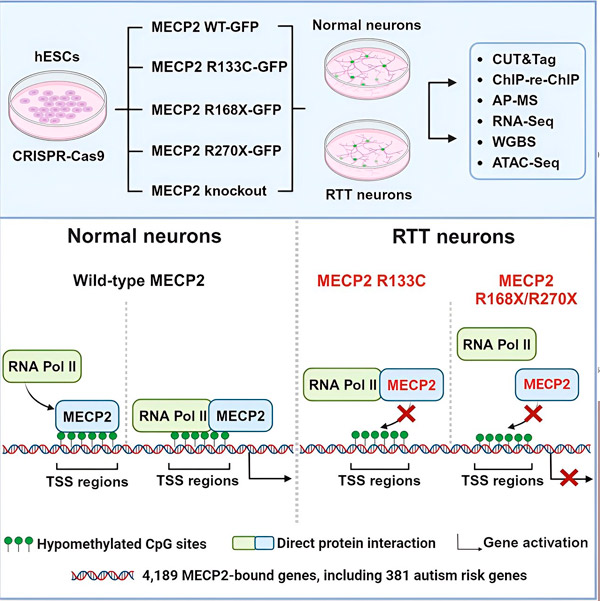
Araştırmacılar ilk olarak MECP2'nin insan nöronal gen dizilerinde, genlerin içinde veya yakınlarındaki DNA'nın düzenleyici bölgelerinde nereye bağlandığına dair detaylı bir harita oluşturdular. DNA ile protein etkileşimlerini yüksek hassasiyetle belirleyebilen CUT&Tag adlı bir yaklaşım kullandılar.
Araştırmacılar, MECP2 ile ilişkili 4.000'den fazla gen buldular. Hastalık durumunda MECP2'nin nerede tükendiğini belirlemek için Rett sendromuyla ilişkili yaygın MECP2 mutasyonlarına sahip nöronlarda haritalamalarını tekrarladılar.
MECP2'nin hangi genlere bağlandığını bilmek, Liu ve Flamier'in MECP2'nin hedefleri ile beyin sağlığı arasında bağlantılar kurmaya başlamasını sağladı. Hedeflerinin çoğunun nöronal aksonların ve sinapsların gelişimi ve işleviyle ilgili olduğunu buldular.
Ayrıca MECP2 hedefleri listesini Simons Vakfı Otizm Araştırma Girişimi'nin (SFARI) otizmle ilişkili genler veri tabanıyla karşılaştırdılar ve bu veri tabanındaki 381 genin MECP2 hedefleri olduğunu buldular.
Kaynak: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Bu bulgular, Rett sendromunda otizm semptomlarının altında yatan mekanizmaların açıklığa kavuşturulmasına yardımcı olabilir ve MECP2'nin otizmdeki olası rolünün araştırılması için iyi bir başlangıç noktası sağlayabilir.
Liu, "Sağlık ve hastalıkta MECP2 epigenomunun ilk entegre haritasını oluşturduk ve bu harita gelecekteki araştırmalara rehberlik edebilir," diyor. "Hangi genlerin MECP2'nin hedefi olduğunu ve hangi genlerin hastalıkta doğrudan bozulduğunu bilmek, Rett sendromunu anlamak ve nöronlardaki gen düzenlemesi hakkında sorular sormak için sağlam bir temel sağlar."
Araştırmacılar ayrıca MECP2'nin hedef genlerinin ifadesini artırıp artırmadığına da baktılar. MECP2'nin bazıları tarafından bir aktivatör, diğerleri tarafından ise bir baskılayıcı olarak tanımlanma geçmişiyle tutarlı olarak, Liu ve Flamier, MECP2'nin her iki rolü de oynadığı örnekler buldular.
Ancak, MECP2 daha çok bir baskılayıcı olarak düşünülürken, Liu ve Flamier bunun çoğunlukla bir aktivatör olduğunu buldular - Jaenisch ve Liu'nun önceki bulgularını doğruladılar. Yeni bir deney, MECP2'nin hedeflerinin en az %80'ini aktive ettiğini gösterdi ve bir diğeri de hedeflerinin %88'ine kadarını aktive ettiğini buldu.
Araştırmacılar tarafından oluşturulan hedef genlerin haritası, MECP2'nin bir aktivatör olarak rolüne dair ek bir içgörü sağladı. MECP2'nin aktive ettiği genler için, tipik olarak genin transkripsiyon başlangıç noktası adı verilen DNA'nın yukarı akışındaki bir bölgesine bağlandığını buldular.
Bu, hücresel makinenin bir genin RNA'ya transkripsiyonu sürecini başlattığı ve ardından RNA'nın gen ifadesinin ürünü olan işlevsel bir proteine çevrildiği yerdir. MECP2'nin gen ifadesinin başladığı transkripsiyon başlangıç noktasında bulunması, gen aktivatörü olarak oynadığı rolle tutarlıdır.
Araştırmacılar daha sonra MECP2'nin gen aktivasyonunda hangi rolü oynadığını belirlemeye koyuldular. MECP2'nin DNA'ya ek olarak bu bölgede hangi moleküllere bağlandığına baktılar ve MECP2'nin RNA polimeraz II (RNA Pol II) adı verilen bir protein kompleksiyle doğrudan etkileşime girdiğini buldular. RNA Pol II, DNA'yı RNA'ya dönüştüren önemli bir hücresel makinedir. RNA Pol II kendi başına genleri bulamaz, bu yüzden işini yapmasına yardımcı olması için çeşitli yardımcı faktörlere veya protein işbirlikçilerine ihtiyaç duyar.
Araştırmacılar, MECP2'nin, RNA Pol II'nin MECP2'nin bağlandığı genlerde transkripsiyonu başlatmasına yardımcı olan böyle bir yardımcı faktör olarak hizmet ettiğini ileri sürüyorlar. MECP2'nin yapısal analizi, molekülün RNA Pol II'ye bağlanan kısımlarını tanımladı ve diğer deneyler, MECP2 kaybının, uygun transkripsiyon başlangıç bölgelerinde RNA Pol II'nin varlığını ve hedef genlerin ifade seviyelerini azalttığını doğruladı.
Bu, Rett sendromunun, MECP2'nin RNA Pol II'ye veya DNA'ya bağlanmasını engelleyen MECP2 mutasyonları nedeniyle MECP2 tarafından hedeflenen genlerin transkripsiyonunun azalmasından kaynaklanabileceğini düşündürmektedir. Bu fikirle tutarlı olarak, hastalıkla ilişkili en yaygın MECP2 mutasyonları, proteinin bir kısmının eksik olduğu ve MECP2 ile RNA Pol II arasındaki etkileşimi değiştirebilen mutasyonlar olan kesilmelerdir.
Araştırmacılar, bulgularının yalnızca MECP2 hakkındaki anlayışımızı değiştirmesini değil, aynı zamanda MECP2'nin beyin gelişimi ve işlevini nasıl etkilediğine dair daha derin ve daha geniş bir anlayışın, Rett sendromu ve otizm de dahil olmak üzere ilgili bozuklukları olan insanlara yardımcı olacak yeni içgörülere yol açabileceğini umuyorlar.
Flamier, "Bu proje, Janisch laboratuvarının işbirlikçi doğasının harika bir örneğidir," diyor. "Rudolf ve ben Rett sendromuyla ilgili belirli bir sorun yaşadık ve sorunu çözebilecek CUT&Tag teknolojisiyle deneyimim vardı. Tartışmalar sonucunda çabalarımızı birleştirebileceğimizi fark ettik ve şimdi MECP2 ve hastalıkla bağlantıları hakkında harika bir bilgi kaynağımız var."